
- Kako bolest fenilketonurija
- Mehanizam razvoja bolesti
- Fenilketonurija u djece
- Simptomi bolesti
- Uzroci i izazovni čimbenici
- dijagnostika
- Liječenje klasične fenilketonurije
- Особенности питания новорожденных и диетотерапия
- Диета для детей дошкольного возраста и школьников
- Группы продуктов при ФКУ
- Kako kontrolirati razinu fenilalanina u krvi
- video
Pomozite razvoju web mjesta, dijelite članak s prijateljima!
Bolest, čija je pojava povezana s manama u genetskom staničnom aparatu, fenilketonuriji, uključena je u nekoliko popisa nasljednih bolesti koje se mogu liječiti. Pionir ove bolesti bio je liječnik iz Norveške I.A. Poslije pada, kasnije je otkriveno da je jedan gen, nazvan gen fenilalanin hidroksilaze (duga ruka 12. kromosoma, koji sadrži do 4, 5% ukupnog DNK materijala stanice), odgovoran za razvoj i tijek bolesti. Naslijeđeni defekt dovodi do djelomične ili potpune deaktivacije jetrenog enzima fenilalanin-4-hidroksilaze.
Kako bolest fenilketonurija
Nasljedna bolest fenilketonurija (PKU) dovodi do kroničnog trovanja organizma toksičnim tvarima koje nastaju uslijed smanjenog metabolizma aminokiselina i procesa fenilalaninske hidroksilacije. Stalna intoksikacija uzrokuje oštećenje središnjeg živčanog sustava (CNS), čija je manifestacija progresivno smanjenje inteligencije (fenilpyruvic oligofrenija).
Bolest sječenja očituje se u prekomjernoj akumulaciji u tijelu fenilalanina i produktima njegovog nepravilnog metabolizma. Drugi čimbenici u razvoju fenilketonurije uključuju oštećenje prijenosa aminokiselina preko krvno-moždane barijere, niske razine neurotransmitera (serotonin, histamin, dopamin). U nedostatku pravovremenog liječenja, bolest dovodi do mentalne retardacije i može uzrokovati smrt djeteta.

Mehanizam razvoja bolesti
Uzročni čimbenik u nastanku poremećaja gena je metabolički blok koji sprječava stvaranje fenilalanin-4-hidroksilaze (enzim odgovoran za pretvorbu aminokiseline fenilalanin u tirozin). Proteinogena aminokiselina tirozin je sastavni dio proteina i pigmenta melanina, stoga je bitan element za funkcioniranje svih tjelesnih sustava, a njegov nedostatak dovodi do fermentopatije.
Posljedica supresije stvaranja metabolita uzrokovanog mutacijskom inaktivacijom enzima je aktiviranje pomoćnih putova za razmjenu fenilalanina. Aromatična alfa-aminokiselina kao rezultat neispravnih metaboličkih procesa raspada se u toksične derivate, koji u normalnim uvjetima nisu formirani:
- Fenilpiruvična kiselina (fenilpiruvat) je masna aromatska alfa-keto kiselina, njezino stvaranje dovodi do mijelinacije neuronskih procesa i demencije;
- Fenil-mliječna kiselina - produkt nastao redukcijom fenilpiruvične kiseline;
- Feniletilamin - početni spoj za biološki aktivne odašiljače elektrokemijskih pulseva, povećava koncentraciju dopamina, adrenalina i noradrenalina;
- orthofhenylacetate je otrovna tvar koja uzrokuje poremećaj metaboličkih procesa masnoća sličnih spojeva u mozgu.
Medicinska statistika sugerira da je patološki izmijenjeni gen prisutan u 2% populacije, ali se ne manifestira. Genetski defekt se prenosi na dijete od roditelja samo ako je bolest prisutna u oba partnera, au 50% slučajeva dijete postaje nositelj mutiranog gena, ostajući zdrav. Vjerojatnost da će fenilketonurija kod novorođenčadi dovesti do bolesti je 25%.
Koji je tip naslijeđen
Bolest sječenja je genetski poremećaj naslijeđen autosomalno recesivno. Ova vrsta nasljeđivanja znači da će se razvoj znakova kongenitalne bolesti pojaviti samo kada dijete naslijedi jedan defektni genokopiju od oba roditelja, koji su heterozigotni nositelji modificiranog gena.
Razvoj kongenitalne bolesti u 99% slučajeva uzrokuje mutaciju gena odgovornog za kodiranje enzima koji osigurava sintezu fenilalanin-4-hidroksilaze (klasična fenilketonurija). Do 1% genetskih bolesti povezano je s mutacijskim promjenama koje se javljaju u drugim genima i uzrokuju nedostatak dihidropteridin reduktaze (PKU tip II) ili tetrahidrobiopterina (PKU tip III).
Fenilketonurija u djece
Klasični oblik genetske bolesti u djece u većini slučajeva očituje se u vanjskim prepoznatljivim znakovima, počevši od 3-9 mjeseci života. Novorođenčad s defektnim genom izgledaju zdravo, a osobitost je specifičan habitus (izgled) djeteta. Ozbiljni simptomi javljaju se 6-12 mjeseci nakon rođenja.
PKU tip II karakterizira činjenica da se prvi klinički simptomi javljaju nakon 1, 5 godine od trenutka rođenja. Simptomi bolesti ne nestaju nakon dijagnoze genetskih abnormalnosti i početka terapije. Ova vrsta urođene bolesti često dovodi do smrti u 2-3 godine života djeteta. Najčešći simptomi tipa II PKU su:
- izražena odstupanja u mentalnom razvoju;
- hiperrefleksija;
- kršenje motoričkih funkcija svih udova;
- sindrom nekontroliranih kontrakcija mišića.
- visok stupanj mentalne retardacije;
- jasno smanjena veličina lubanje u odnosu na druge dijelove tijela;
- spastičnost mišića (uz moguću potpunu nepokretnost udova).

Manifestacije bolesti sječe
Tijekom kliničkih ispitivanja i promatranja, sugerirano je da učinci toksičnih derivata metabolizma fenilalanina uzrokuju smanjenje intelektualnih sposobnosti, što je progresivno i može dovesti do demencije (oligofrenija, idiotizam). Među navodnim uzrocima ireverzibilnih poremećaja moždane aktivnosti, najpouzdaniji je nedostatak neurotransmitera koji prenose impulse između neurona uzrokovanih smanjenjem razine tirozina.
Točna uzročna veza između nasljedne bolesti i poremećaja mozga još nije identificirana, kao ni mehanizam razvoja zbog fenilketonurije takvih mentalnih stanja kao što su ekopraksija, eholalija, napadi ljutnje i razdražljivosti. Podaci iz rezultata ispitivanja pokazuju da fenilalanin ima izravan toksični učinak na mozak, što također može uzrokovati smanjenje inteligencije.
Tijelo i fenotipske značajke
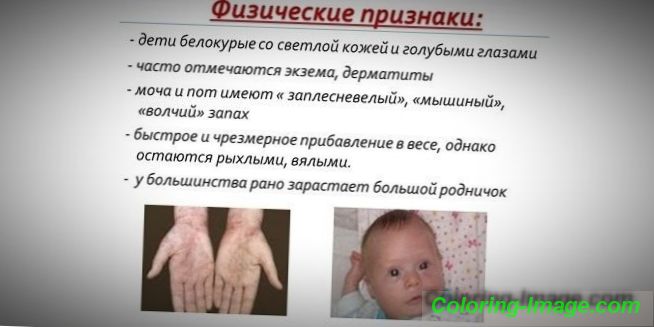
Zbog činjenice da pigment kože i kose ovisi o razini tirozina u mitohondrijama hepatocita, a fenilketonurija zaustavlja konverziju fenilalanina, bolesnici s ovom bolešću imaju fenotipske značajke (recesivni simptomi). Povećani tonus mišića uzrokuje odstupanje u tijelu - postaje displastično. Različiti vanjski znakovi fenilketonurija uključuju:
- hipopigmentacija - svijetla koža, blijedoplave oči, izbijeljena kosa;
- plavetnila udova;
- smanjena veličina glave;
- određeni položaj tijela - kada pokušavate stajati ili sjediti, dijete preuzima poziciju "krojača" (ruke i noge savijene u zglobovima).
Simptomi bolesti
S pravovremenim otkrivanjem, Fellingova bolest pogodna je za uspješno liječenje prilagodbom prehrane, a razvoj djeteta odvija se u skladu s njegovom dobnom skupinom. Poteškoća u otkrivanju mutacije gena je u tome što je teško otkriti rane znakove čak i za iskusnog pedijatra. Težina simptoma kongenitalne bolesti raste kako dijete raste, jer uporaba proteinske hrane doprinosi razvoju CNS poremećaja.
Znakovi kod novorođenčadi
Tijekom prvih dana života djeteta, teško je otkriti znakove patoloških abnormalnosti - dijete se ponaša prirodno i nema razvojnog kašnjenja. Simptomi bolesti po prvi put počinju se pojavljivati 2-6 mjeseci nakon rođenja. Roditelje treba upozoriti na ponašanje djeteta koje karakterizira niska aktivnost, letargija ili, obratno, anksioznost, hiper razdražljivost.
Početkom dojenja, proteini počinju ulaziti u tijelo novorođenčadi s mlijekom, što služi kao katalizator za pojavu prvih znakova koji jasno pokazuju da je bolest počela napredovati. Specifične kliničke manifestacije bolesti uključuju:
- trajno povraćanje (često se pogrešno shvaća za prirođeno suženje pylorusa);
- česta regurgitacija;
- nedostatak odgovora na vanjske podražaje;
- mišićna distonija (smanjena napetost mišića);
- konvulzivni sindrom (epileptički ili neepileptički napadi).
Simptomi u djece nakon 6 mjeseci
Ako se manifestacija genetske bolesti nije pojavila (ili nije primijećena) tijekom prvih 6 mjeseci od trenutka rođenja djeteta, nakon tog razdoblja već je moguće točno odrediti psihomotorni lag. Simptomi genetskih poremećaja uzrokovanih nedostatkom enzima kod djece starije od šest mjeseci su:
- smanjenje aktivnosti (do potpune indiferentnosti);
- bez pokušaja da ustanu sami;
- poseban "mišji" miris kože (miris plijesni nastaje zbog izlučivanja toksičnih derivata fenilalanina kroz žlijezde znoja i urina);
- gubitak sposobnosti vizualnog prepoznavanja roditelja;
- ljuštenje kože;
- pojavu dermatitisa, ekcema, skleroderme.
Napredovanje bolesti u odsutnosti liječenja u djetinjstvu
Ako razvojna oštećenja nisu identificirana u ranom djetinjstvu, a odgovarajuće liječenje nije provedeno, tada bolest počinje aktivno napredovati i često dovodi do invaliditeta. Nedostatak terapije u ranom stadiju bolesti uzrokuje pojavu sljedećih simptoma bolesti već u dobi od 1, 5 godina:
- mikrocefalija (smanjena veličina mozga);
- prognatija (pomicanje gornje denticije prema naprijed);
- kasni zubi;
- hipoplazija cakline (stanjivanje ili potpuna odsutnost zubne cakline);
- odgođeni razvoj govora do potpunog nedostatka govora;
- 3, 4 stupanj oligofrenije (mentalna retardacija, mentalna retardacija);
- kongenitalne srčane mane (defekti u strukturi srčanog mišića, dijelova srca, velikih krvnih žila);
- poremećaji vegetativnog sustava (akrocijanoza, prekomjerno znojenje, arterijska hipotenzija);
- zatvor.
Uzroci i izazovni čimbenici
Za mutacije s autosomno recesivnim obrascem nasljeđivanja, defektni gen mora biti naslijeđen od oba roditelja. Genetske bolesti ovog tipa javljaju se s istom učestalošću kod novorođenčadi i djevojčica. Patogeneza PKU određena je metaboličkim poremećajem fenilalanina, koji se može pojaviti u 3 oblika. Samo klasična fenilketonurija tipa I može se liječiti dijetalnom terapijom.
Atipični oblici bolesti ne mogu se izliječiti podešavanjem prehrane. Ta su odstupanja uzrokovana nedostatkom tetrahidropterina, dehidropterin reduktaze (rjeđe piruvoil tetrahidropterin sintetaza, gvanozin-5-trifosfat ciklohidrolaza, itd.). Većina slučajeva smrtnih ishoda zabilježena je kod bolesnika s rijetkim varijacijama PKU, dok su kliničke manifestacije svih oblika bolesti slične. Rizik dobivanja djeteta s mutiranim genom fenilalanin hidroksilaze povećava se ako su njegovi roditelji bliski srodnici (u usko povezanim brakovima).
dijagnostika
Ako se sumnja na genetski poremećaj, postavlja se dijagnoza na temelju kombinacije podataka dobivenih proučavanjem povijesti bolesti - genealoške informacije, rezultati kliničkih i medicinskih genetskih istraživanja. Za pravodobno otkrivanje urođenih bolesti (PKU, cistična fibroza, galaktosemija, itd.) Razvijen je obvezni program masenog probira za sve novorođenčad (neonatalni probir).
Ako budući roditelji znaju za prijevoz mutiranog gena, moderna medicina nudi načine otkrivanja defekta tijekom trudnoće (prenatalna dijagnoza fetusa invazivnom metodom). Za odvajanje fenilketonurije u vrste prema težini koristi se uvjetna klasifikacija koja se temelji na razini fenilalanina u tekućini bez fibrina dobivenoj iz krvne plazme:
- Teška fenilketonurija - 1200 µmol / l.
- Prosjek je 60-1200 μmol / l.
- Svjetlo (ne zahtijeva liječenje) - 480 µmol / l.
Test probira
Identifikacija genetskih abnormalnosti događa se u nekoliko faza. U prvoj fazi u rodilištu, za sve bebe u 3-5 dana života, periferna krv se uzima iz pete za istraživanje. Materijal se nanosi na papirnati oblik i šalje u biokemijski laboratorij, gdje je biokemijska analiza. U drugom stupnju probirnog testa utvrđeno je da je koncentracija fenilalanina normalna.
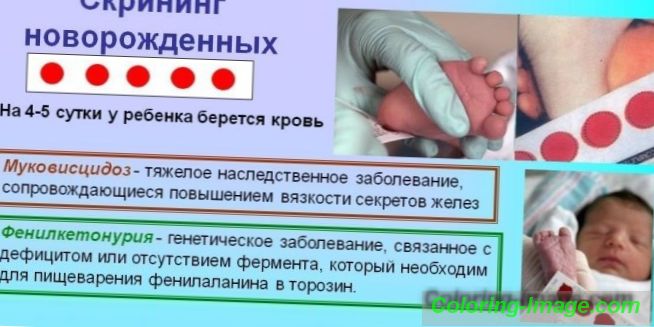
Ako se ne otkriju patološke promjene, dijagnoza je dovršena i unosi se u iskaznicu djeteta. U slučaju odstupanja od norme, rezultati dijagnoze šalju se pedijatru kako bi se osigurao pregled uzorka krvi novorođenčeta. Zdravlje djeteta ovisi o pravovremenoj i točnoj provedbi svih mjera za utvrđivanje odstupanja. Ako se dijagnoza potvrdi nakon ponovnog pregleda, roditelji djeteta bit će upućeni na kliniku za dječju genetiku u svrhu liječenja.
Analize i studije koje potvrđuju dijagnozu
Ponovljena dijagnostika kod otkrivanja abnormalnosti tijekom početnog probirnog testa provodi se ponovljenim testovima. Uz određivanje sadržaja fenilalanina u krvi, metode dijagnosticiranja PKU u djece i odraslih uključuju:
- Ispitivanje paležom - određivanje fenilpiruvične kiseline u mokraći dodavanjem željeznog klorida u biomaterijal (javlja se plavo-zeleno bojenje);
- Guthrie test - procjena stupnja reakcije mikroorganizama na metaboličke produkte ili enzime sadržane u krvi pacijenta;
- kromatografija - proučavanje kemijskih svojstava tvari raspodijeljenih između dvije faze;
- fluorimetrija - zračenje biomaterijala s monokromatskim zračenjem za određivanje koncentracije tvari koje se u njemu nalaze;
- elektroencefalografija - dijagnostika električne aktivnosti mozga;
- magnetska rezonancija je ekscitacija atomskih jezgri stanica elektromagnetskim valovima i mjerenje njihovog odgovora.
Liječenje klasične fenilketonurije
Osnova terapije fenilketonurijom je ograničenje potrošnje proizvoda koji su izvor životinjskih i biljnih proteina. Jedina metoda uspješnog liječenja je dijetalna terapija, čija adekvatnost se procjenjuje prema sadržaju fenilalanina u krvnom serumu. Najveća dopuštena razina aminokiselina u bolesnika različitih dobnih skupina je:
- kod novorođenčadi i djece do 3 godine - do 242 µmol / l;
- kod predškolske djece - do 360 µmol / l;
- u bolesnika u dobi od 7 do 14 godina - do 480 µmol / l;
- u adolescenata, do 600 μmol / l.
Djelotvornost prehrane ovisi o fazi u kojoj je bolest prilagođena. U ranoj dijagnozi kongenitalne patologije propisana je dijetalna terapija od 8. tjedna života (nakon tog razdoblja počinju nepovratne promjene). Nedostatak pravodobnih mjera dovodi do komplikacija i smanjenja razine inteligencije za 4 boda za 1 mjesec od trenutka rođenja do početka liječenja.

Zbog činjenice da terapijska dijeta za fenilketonuriju podrazumijeva potpunu eliminaciju životinjskih bjelančevina iz prehrane, potrebno je koristiti i druge izvore esencijalnih aminokiselina, kao i vitamine skupine B, kalcijeve i fosforne mineralne spojeve. Proizvodi označeni kao dodaci prehrani bez proteina uključuju:
- proteinski hidrolizati (Amigen, Aminazol, Fibrinosol);
- mješavina bez fenilalanina zasićena esencijalnim aminokiselinama - tetrafen, fenil slobodna.
Uz korektivne mjere za otklanjanje uzroka narušenog funkcioniranja tijela, potrebno je provesti simptomatsko liječenje s ciljem otklanjanja oštećenja govora i normalizacije motoričke koordinacije. Kombinirana terapija uključuje fizioterapeutske postupke, masažu, pomoć logopeda, psihologa, gimnastičke vježbe. U nekim slučajevima, uz dijetalnu terapiju, indicirana je uporaba antikonvulziva, nootropnih i vaskularnih lijekova.
Značajke liječenja atipičnih oblika
Fenilketonurija tipa II i tipa III ne može se liječiti dijetom s niskim sadržajem proteina - razina fenilalanina u krvi ostaje nepromijenjena kada je unos proteina u organizam ograničen ili klinički simptomi napreduju čak i sa smanjenjem razine aminokiselina. Učinkovita terapija ovih oblika bolesti provodi se pomoću:
- tetrahidrobiopterin - faktor zahvaćenog enzima;
- sintetski analozi tetrahidrobiopterina - te tvari bolje prodiru u krvno-moždanu barijeru;
- препаратов заместительной терапии – не устраняют причину фенилкетонурии, но поддерживают нормальное функционирование организма (Леводопа совместно с Карбидофой, 5-окситриптофан, 5-формилтетрагидрофолат);
- гепатопротекторов – поддерживают функционирование печени;
- antikonvulzivi;
- введения гена фенилаланингидроксилазы в печень – экспериментальный метод.
Особенности питания новорожденных и диетотерапия
На первом году жизни ребенка с ФКУ допустимо кормление грудным молоком, но его количество должно быть ограничено. До 6 месяцев допустимым уровнем потребления фенилаланина является 60-90 мг на 1 кг веса малыша (в 100 г молока содержится 5, 6 мг фенилаланина). Начиная с 3 месяцев, рацион ребенка следует поэтапно расширять, вводя в него фруктовые соки и пюре.
Детям с 6 месяцев разрешено введение в рацион овощных пюре, каш (из саго), безбелковых киселей. После 7 месяцев можно давать малышу низкобелковые макаронные изделия, с 8 месяцев – хлеб, не содержащий белка. Возраст, до которого следует ограничивать поступление белка в организм больного ребенка, не установлен. Врачи до настоящего времени дискутируют по вопросу целесообразности пожизненной диетотерапии, но сходятся во мнении, что минимум до 18 лет необходимо придерживаться диетического питания.
Фенилкетонурия, диагностированная у женщины, не является поводом отказаться от рождения ребенка. Будущим мамам с ФКУ для предупреждения повреждения плода во время беременности и профилактики возможных осложнений необходимо до планируемого зачатия и во время вынашивания ребенка придерживаться диеты с ограничением фенилаланина (его уровень в крови должен быть до 242 мкмоль/л).
Безлактозные смеси для малышей
Диета при фенилкетонурии базируется на существенном уменьшении дозы натурального белка в суточном рационе, но организм новорожденного ребенка не может развиваться нормально при отсутствии необходимых микроэлементов. Для восполнения потребности малыша в белке применяются безлактозные аминокислотные смеси, которыми, согласно российскому законодательству, больные должны обеспечиваться бесплатно.
Толерантность грудничка к фенилаланину в течение первого года жизни стремительно изменяется, поэтому необходимо контролировать его концентрацию в крови ребенка и вносить корректировки в диету. Смеси рассчитаны на определенные возрастные группы:
- малышам до года назначаются Афенилак 15, Аналог-СП, PKU-1, PKU-mix, PKU Anamix;
- детям, старше 1 года, назначают обогащенные витаминами и минералами смеси с повышенным содержанием белка – PKU Prima, P-AM Universal, ФКУ-1, ФКУ-2, ХР Максамейд, ХР Максамум.

Диетические продукты для пополнения запасов белка
Одним из основных компонентов пищевой диеты при фенилкетонурии являются малобелковые продукты на основе крахмала. Эти добавки содержат гидролизат казеина, триптофан, тирозин, метионин, азот и обеспечивают суточную потребность организма ребенка в белке, который необходим для нормального развития и роста. Специализированными продуктами, восполняющими нехватку необходимых минералов и аминокислот при их нехватке в рационе питания, являются:
- Берлофен;
- Циморган;
- Минафен;
- Апонти.
Диета для детей дошкольного возраста и школьников
По мере адаптации организма к фенилаланину детям с возраста 5 лет можно постепенно уменьшать ограничения в питании. Расширение рациона происходит путем введения круп, молочных продуктов, мясных изделий. Школьники старших классов имеют уже высокую толерантность к фенилаланину, поэтому в этом возрасте можно продолжить расширение диеты, при этом необходимо отслеживать реакцию на все изменения в питании. Для контроля состояния ребенка применяются следующие способы:
- оценка неврологических показателей, психологического состояния;
- контроль показателей электроэнцефалограммы;
- определение уровня фенилаланина.
Группы продуктов при ФКУ
В рацион питания пациентов с ФКУ наряду с малобелковыми крахмалистыми продуктами и лечебными смесями входят и продукты натурального происхождения. При составлении меню следует четко рассчитывать количество потребляемого белка и не превышать рекомендованную врачом дозировку. Для исключения токсического влияния на организм разработаны 3 списка продуктов, которые содержат запрещенные (красный), нерекомендованные (оранжевый) и разрешенные (зеленый) позиции.
Красный список
Фенилкетонурия развивается на фоне отсутствия фермента, превращающего фенилаланин в тирозин, поэтому высокое содержание белка является поводом для отнесения продуктов в запрещенный (красный) список. Позиции из этого перечня следует полностью исключить рациона питания больного ФКУ:
- meso;
- unutarnji organi životinja, iznutrice;
- kobasice, kobasice;
- plodovi mora (uključujući ribe);
- jaja svih ptica;
- fermentirani mliječni proizvodi;
- matice;
- plodovi mahunarki i žitarica;
- proizvodi od soje;
- Posude koje sadrže želatinu;
- slastice;
- aspartam.
Narančasti popis
Proizvodi koji treba dozirati u tijelo djeteta s dijagnozom PKU uključeni su u narančasti popis. Uključivanje u prehranu pozicija s ovog popisa je prihvatljivo, ali u strogo ograničenim količinama. Iako ti proizvodi ne sadrže mnogo bjelančevina, ja također mogu povećati razinu fenilalanina, pa se njihova uporaba ne preporučuje:
- konzervirano povrće;
- jela od krumpira i riže;
- kupus;
- mlijeko;
- šerbet.
Zeleni popis
Proizvodi bez proteina dopušteni su za uporabu u bolesnika s dijagnozom fenilketonurije bez ograničenja. Prije kupnje predmeta s zelene liste, morate pregledati sastav prikazan na pakiranju i provjeriti da nema aspartam boje koja sadrži fenilalanin:
- voće;
- povrće (osim krumpira i kupusa);
- bobice;
- zeleno;
- škrobaste žitarice (sago);
- med, šećer, džem;
- brašno od kukuruznog ili rižinog brašna;
- ulja, masti (kremasto, povrće, masline).
Kako kontrolirati razinu fenilalanina u krvi
Fenilketonurija je neizlječiva bolest koja se može pretvoriti u fazu stagnacije korištenjem dijetetske terapije i terapijskih mjera. Promjenom životnih uvjeta, pothranjenošću, bolest se može ponovno pogoršati, pa pacijenti trebaju doživotno promatranje. Postupak praćenja sastoji se u periodičnom određivanju razine fenilalanina u krvi. Učestalost testiranja ovisi o dobi pacijenta:
- do 3 mjeseca - provjeru krvi treba provoditi tjedno dok se ne postignu stabilni rezultati;
- od 3 mjeseca do 1 godine - 1-2 puta mjesečno;
- od 1 do 3 godine - 1 put u 2 mjeseca;
- stariji od 3 godine - tromjesečno.

Krv za testiranje se uzima 3-4 sata nakon obroka. Osim screeninga, praćenje PKU prati se utvrđivanjem nutritivnog statusa, fizičkog, emocionalnog razvoja pacijenta, razine intelektualnih sposobnosti i jezičnog razvoja. Prema rezultatima opažanja, možda će biti potrebno napraviti dodatnu dijagnostiku uz sudjelovanje relevantnih stručnjaka.